
Esclerose sistêmica é uma doença autoimune sistêmica multifacetada e uma das mais complexas doenças do tecido conjuntivo. Dentre as doenças reumáticas autoimunes, é a que apresenta maior mortalidade caso-específica, onde mais da metade dos pacientes morrem por causas diretamente relacionadas à doença (na maioria das vezes por complicações cardiorrespiratórias)1,2.
A ampla gama de manifestações clínicas, ainda que não ameaçadores à vida, como úlceras digitais e alterações do trato digestivo, impacta diretamente na função e a qualidade de vida do paciente, evidenciando a importante morbidade determinada pela doença. Esse pleomorfismo clínico é resultante de sua complexa patogênese, que envolve três pilares que interagem entre si: a inflamação autoimune mediada, a vasculopatia (dano microvascular) e a fibrose (ativação de fibroblastos). Em espectro mais amplo, esse pleomorfismo, que envolve patogênese, fenótipos e manifestações clínicas, dificulta a realização de estudos clínicos e a obtenção de terapias efetivas1.
O atual conhecimento sobre os mecanismos patogênicos da esclerose sistêmica evidencia que ela não pode mais ser considerada uma doença crônica com curso indolente, que evolui até certo ponto e depois regride ou fica estagnada; ao contrário, como outras doenças reumáticas, evolui com fases de remissão e atividade. Estudos observacionais demonstraram que, na esclerose sistêmica difusa, os processos patogênicos que acometem os órgãos internos ocorrem nos primeiros três anos de doença1,4. Assim, o diagnóstico precoce e a intervenção imediata são fundamentais para parar ou prevenir a progressão da doença e minimizar os danos.
As novas perspectivas para a abordagem da esclerose sistêmica levam em consideração as necessidades não atendidas como:
-
- Diagnóstico muito precoce – O grupo europeu de estudo e pesquisa em esclerose sistêmica (EUSTAR) realizou um consenso Delphi para identificar pacientes com doença subclínica (estudo VEDOSS). Os objetivos eram diagnóstico e tratamento precoces, antes do desenvolvimento de fibrose e dano (janela de oportunidade). Três sinais de alerta foram propostos: fenômeno de Raynaud (FRy), “puffy fingers”/esclerodactilia e ANA reagente. Caso esses três sinais estivessem presentes, prosseguia-se com a realização de capilaroscopia e pesquisa de autoanticorpos específicos (anticentrômero e anti Scl70)5,6. Um estudo prévio com cerca de 600 pacientes (sem doença do tecido conjuntivo) para avaliação do FRy demonstrou que 73% desses pacientes, que apresentaram alterações à capilaroscopia e autoanticorpo específico reagente, desenvolveram esclerose sistêmica após dez anos, enquanto 98% dos que não apresentavam alterações à capilaroscopia nem autoanticorpos específicos reagentes permaneceram como FRy primário7.
- Medicina personalizada – Tratar o doente e não a doença. Avanços nos campos de tecnologia e bioinformática possibilitaram a análise de centenas a milhares de genes, seus produtos de transcrição (como RNAs codificadores e não codificadores) e proteínas correspondentes em grandes coortes de pacientes, proporcionando a análise integrativa de variações genéticas individuais8,9. A análise de fibroblastos a partir de biopsia cutânea em pacientes com esclerose sistêmica permitiu a identificação de quatro tipos de perfis de expressão genética: proliferativo, inflamatório, limitado e semelhante ao normal8,10-13. Cada grupo tem uma assinatura de expressão genética única e se correlaciona com manifestações clínicas variáveis. Por exemplo, o subgrupo proliferativo apresenta comprometimento cutâneo mais severo, enquanto o subgrupo limitado apresenta maior severidade do FRy13,14. A identificação do perfil de expressão genética possibilita medicina personalizada com terapêutica individualizada. Indivíduos do subgrupo proliferativo respondem melhor a medicamentos antifibróticos (como inibidores da tirosina quinase e fresolimumabe) e indivíduos do subgrupo inflamatório, a imunossupressores (como micofenolato mofetil, rituximabe, tocilizumabe e abatacept). Já indivíduos dos subgrupos limitado e semelhante ao normal parecem ter melhor prognóstico e respondem a diversos tipos de tratamentos,10,15.
- Medicina de precisão – O conceito de terapia-alvo pode ter diferentes significados na esclerose sistêmica. Pode considerar o tratamento de órgãos específicos (como rins ou pulmões) ou sintomas específicos (como FRy ou refluxo gastroesofágico), ou ainda pode se referir ao tratamento de mecanismos específicos da doença, como ativação imune, inflamação, vasculopatia ou fibrose. “A conotação mais específica para terapia-alvo seria a atenuação ou estimulação de um subtipo celular, molécula ou via intracelular, que impactaria múltiplos processos na patogênese da doença”2. O tratamento atual é não curativo e é frequentemente baseado no órgão acometido. Entretanto, existem novas terapias potencialmente modificadoras da doença em estudo. Essas terapias são decorrentes da melhor compreensão das vias patogênicas-chave e têm como alvo seus diferentes mecanismos (vasculopatia, inflamação/desregulação imune e fibrose)16.
Quadro 1. Novas terapias potenciais em esclerose sistêmica.
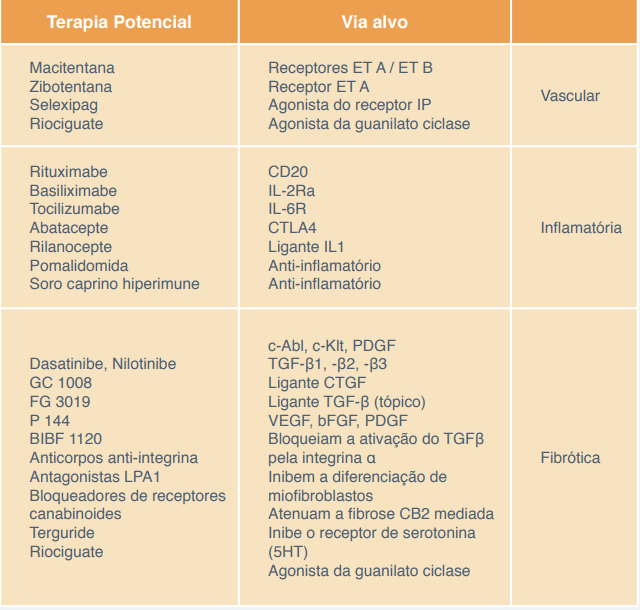
Fonte: Nagaraja, Denton e Khanna (2015)16.
-
-
-
- Métodos (biomarcadores) para avaliação da atividade e progressão da doença e para avaliação da resposta terapêutica – Biomarcadores são definidos como um parâmetro fisiológico ou macromoléculas biológicas que podem ser objetivamente medidos para servir como indicador ou marcador tanto de uma via normal como de uma via patogênica. Eles podem ser transversais (utilizados para auxiliar no diagnóstico e estadiamento de doenças), longitudinais (úteis para avaliar atividade, progressão e prognóstico de doenças) ou farmacodinâmicos (utilizados para avaliar resposta terapêutica)8.
-
-
4.1 Biomarcadores atuais
Os biomarcadores disponíveis atualmente para diagnóstico e/ou acompanhamento dos pacientes com esclerose sistêmica são: escore cutâneo modificado de Rodnan (mRSS), autoanticorpos (ANA, ACA, antiScl 70 e anti RNA polimerase III) e a capilaroscopia. O mRSS consiste na avaliação subjetiva da induração cutânea e é atualmente a única medida de resultado validada para uso na prática clínica e nos estudos clínicos8. Quanto aos autoanticorpos, estudos demonstraram que a positividade e o padrão do ANA têm valor prognóstico em pacientes com esclerose sistêmica subclínica ou muito precoce17-20. Em outro estudo, o padrão nucleolar foi associado à rápida progressão da doença20.O ACA foi associado à menor mortalidade e à menor incidência de fibrose pulmonar e crise renal, enquanto o anti Scl 70 foi associado à maior mortalidade e à maior incidência de fibrose pulmonar21. O anti RNA polimerase III foi associado à maior incidência de crise renal e a tumores malignos22. Já a capilaroscopia pode ser utilizada para diagnóstico e acompanhamento dos pacientes com esclerose sistêmica. São descritos três padrões distintos, específicos para esclerose sistêmica: inicial (dilatação das alças capilares, sem desvascularização), ativo (dilatação e micro-hemorragias, pouca desvascularização) e tardio (desvascularização importante e ramificação das alças capilares)23-25.
4.2 Novos e futuros biomarcadores Vesículas extracelulares (EVs): coleção não homogênea de estruturas ligadas à membrana plasmática que são secretadas por todos os tipos celulares8.
-
-
-
- Tanscriptomas: permitem definir padrões de expressão genética8. Estudos em pacientes com esclerose sistêmica demonstraram padrões expressos em fibroblastos cutâneos classificados como proliferativo, inflamatório, limitado e semelhante ao normal13 e em fibroblastos de outros tecidos (pulmão e esôfago) classificados como inflamatório e proliferativo26-28.
- Micro RNAs: família de RNAs curtos (18 nucleotídeos a 23 nucleotídeos) não codificadores. Sua expressão anormal parece estar associada a algumas doenças autoimunes, como esclerose sistêmica, lúpus e artrite reumatoide. São encontrados no sangue, na urina, em lágrimas e no leite materno, sendo facilmente acessíveis à coleta. Nos últimos anos, vários estudos demonstraram que os níveis dessas moléculas estão alterados no soro de pacientes com esclerose sistêmica. A expressão dos miRNAs 150, 30b, 196a e let-7a está reduzida nos fibroblastos da pele de pacientes com esclerose sistêmica e se associa à severidade do acometimento cutâneo. Já os miRNAs 21 e 155 são considerados pró-fibróticos e têm sua expressão aumentada nos fibroblastos cutâneos de pacientes com esclerose sistêmica8,29.
-
-
Mesmo em face dos avanços que ocorreram nos últimos anos no conhecimento de sua patogênese, a esclerose sistêmica persiste como uma doença desafiadora. É necessário assim utilizar o arsenal disponível para a realização de um diagnóstico cada vez mais precoce e a instituição de tratamento adequado. Bem como é fundamental reconhecer e priorizar as necessidades não atendidas, pois a atenção voltada para estas conduz, progressivamente, ao objetivo principal, que é o controle da doença e a obtenção de uma qualidade de vida adequada.
Referências
-
-
-
- DEL PAPA, N.; ZACCARA, E. From mechanisms of action to therapeutic application: A review on current therapeutic approaches and future directions in systemic sclerosis. Best Practice & Research Clinical Rheumatol., v. 29, p. 756-769, 2015.
- DENTON, C. P. Systemic sclerosis: from pathogenesis to targeted therapy. Clin. Exp. Rheumatol., v. 33, suppl. 92, S 3-7, 2015.
- BARSOTTI, S.; STAGNARO, C.; DELLA ROSSA, A. Systemic sclerosis: a critical digest of the recent literature. Clin. Exp. Rheumatol., v. 33, suppl. 91, S 3-14, 2015.
- STEEN, V. D.; MEDSGER, T. A. Jr. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis and Rheumatism, v. 43, n. 11, p. 2437-2444, 2000.
- ELHAI, M. et al. Systemic sclerosis: recent insights. Joint Bone Spine, v. 82, p. 148-153, 2015.
- AVOUAC, J. et al. Preliminary criteria for the very early diagnosis of systemic sclerosis: results of a Delphi Consensus Study from EULAR Scleroderma Trials and Research Group. Ann. Rheum. Dis., v. 70, p. 476-481, 2011.
- KOENIG, M. et al. Autoantibodies and microvascular damage are independent predictive factors for the progression of Raynaud’s phenomenon to systemic sclerosis: a twenty-year prospective study of 586 patients, with validation of proposed criteria for early systemic sclerosis. Arthritis Rheum., v. 58, p. 3902-3912, 2008.
- WERMUTH, P. J. et al. Existing and novel biomarkers for precision medicine in systemic sclerosis. Nature Reviews Rheumatology, v. 14, p. 421-432, 2018.
- LAUFER, V. A. et al. Integrative approaches to understanding the pathogenic role of genetic variation in rheumatic diseases. Rheum. Dis. Clin. North Am., v. 43, p. 449-466, 2017.
- ZUO, X. et al. Systematic approach to understanding the pathogenesis of systemic sclerosis. Clinical Genetics, v. 92, p. 365-371, 2017.
- WHITFIELD, M. L. et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc. Natl. Acad. Sci. USA, v. 100, p. 12319-12324, 2003.
- GARDNER, H. et al. Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum., v. 54, p. 1961-1973, 2006.
- MILANO, A. et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One, v. 3, e 2696, 2008.
- ASSASSI, S. et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol., v. 67, p. 3016-3026, 2015.
- TARONI, J. N. et al. A functional genomic meta-analysis of clinical trials in systemic sclerosis: toward precision medicine and combination therapy. J. Clin. Invest. Dermatol., v. 137, p. 1033-1041, 2017.
- NAGARAJA, V.; DENTON, C. P.; KHANNA, D. New pathways in the pathogenesis of SSc – Old medications and new targeted therapies in systemic sclerosis. Rheumatology, v. 54, n. 11, p. 1944-1953, 2015.
- DOMSIC, R. T. Scleroderma: the role of serum autoantibodies in defining specific clinical phenotypes and organ system involvement. Curr. Opin. Rheumatol., v. 26, p. 646-652, 2014.
- SIROTTI, S. et al. Personalized medicine in rheumatology: the paradigm of serum autoantibodies. Auto. Immun. Highlights, v. 8, p. 10, 2017.
- MUELLER, M. et al. Relation of nailfold capillaries and autoantibodies to mortality in patients with Raynaud phenomenon. Circulation, v. 133, p. 509-517, 2016.
- SULLI, A. et al. Progression of nailfold microvascular damage and antinuclear antibody pattern in systemic sclerosis. J. Rheumatol., v. 40 p. 634-39, 2013.
- NIHTYANOVA, S. I. et al. A Practical classification of systemic sclerosis using subset and autoantibodies for the purpose of early risk stratification [abstract]. Arthritis Rheumatol., v. 70, suppl. 10, 2018. Disponível em: <https://acrabstracts.org/abstract/a-practical-classification-of-systemic-sclerosis-using-subset-and-autoantibodies-for-the-purpose-of-early-risk-stratification>. Acesso em: 28 jan. 2019.
- XU, G. J. et al. Systemic autoantigen analysis identifies a distinct subset of scleroderma with coincident cancer. Proc. Natl. Acad. Sci. USA, v. 113, p. 57526-57534, 2016.
- MARICQ, H. R. et al. Diagnostic potential of in vivo microscopy in scleroderma and related disorders. Arthritis Rheum., v. 23, p. 183-189, 1980.
- MARICQ, H. R., WEINBERGER, A. B.; LEROY, E. C. Early detection of scleroderma-spectrum disorders by in vivo capillary microscopy: a prospective study of patients with Raynaud’s phenomenon. J. Rheumatol., v. 9, p. 289-291, 1983.
- CUTOLO, M. et al. Assessing microvascular changes in systemic sclerosis diagnosis and management. Nat. Rev. Rheumatol., v. 6, p. 578-587, 2010.
- PENDERGRASS, S. A. et al. Limited systemic sclerosis patients with pulmonary arterial hypertension show biomarkers of inflammation and vascular injury. PLoS ONE, v. 5, 2010.
- LENNA, S. et al. Increased expression of endoplasmic reticulum stress and unfolded protein response genes in peripheral blood mononuclear cells from patients with limited cutaneous systemic sclerosis and pulmonary arterial hypertension. Arthritis Rheum., v. 65, p. 1357-1366, 2013.
- TARONI, J. N. et al. Molecular characterization of systemic sclerosis esophageal pathology identifies inflammatory and proliferative signatures. Arthritis Res. Ther., v. 17, p. 194, 2015.
- WALCZYK, M.; PARADOWSKA-GORYCKA, A.; OLESINSKA, M. Epigenetics: The future direction in Systemic Sclerosis. Scandinavian Journal of Immunology, v. 86, p. 427-435, 2017.
-
-

Autora: Dra. Rossana de Moura Guedes
Reumatologista pela Universidade Federal de Goiás e Sociedade Brasileira de Reumatologia
Professora da Pontifícia Universidade Católica de Goiás e da Universidade de Rio Verde (Campus Aparecida)
Preceptora da Residência de Reumatologia do Hospital Geral de Goiânia.